 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: [email protected]. Type 508 Accommodation and the title of the report in the subject line of e-mail. Surveillance for Safety After Immunization: Vaccine Adverse Event Reporting System (VAERS) --- United States, 1991--2001Please note: An erratum has been published for this article. To view the erratum, please click here. Weigong Zhou, M.D., Ph.D.1, 2 Abstract Problem/Condition: Vaccines are usually administered to healthy persons who have substantial expectations for the safety of the vaccines. Adverse events after vaccinations occur but are generally rare. Some adverse events are unlikely to be detected in prelicensure clinical trials because of their low frequency, the limited numbers of enrolled subjects, and other study limitations. Therefore, postmarketing monitoring of adverse events after vaccinations is essential. The cornerstone of monitoring safety is review and analysis of spontaneously reported adverse events. Reporting Period Covered: This report summarizes the adverse events reported to the Vaccine Adverse Event Reporting System (VAERS) from January 1, 1991, through December 31, 2001. Description of Systems: VAERS was established in 1990 under the joint administration of CDC and the Food and Drug Administration (FDA) to accept reports of suspected adverse events after administration of any vaccine licensed in the United States. VAERS is a passive surveillance system: reports of events are voluntarily submitted by those who experience them, their caregivers, or others. Passive surveillance systems (e.g., VAERS) are subject to multiple limitations, including underreporting, reporting of temporal associations or unconfirmed diagnoses, and lack of denominator data and unbiased comparison groups. Because of these limitations, determining causal associations between vaccines and adverse events from VAERS reports is usually not possible. Vaccine safety concerns identified through adverse event monitoring nearly always require confirmation using an epidemiologic or other (e.g., laboratory) study. Reports may be submitted by anyone suspecting that an adverse event might have been caused by vaccination and are usually submitted by mail or fax. A web-based electronic reporting system has recently become available. Information from the reports is entered into the VAERS database, and new reports are analyzed weekly. VAERS data stripped of personal identifiers can be reviewed by the public by accessing http://www.vaers.org. The objectives of VAERS are to 1) detect new, unusual, or rare vaccine adverse events; 2) monitor increases in known adverse events; 3) determine patient risk factors for particular types of adverse events; 4) identify vaccine lots with increased numbers or types of reported adverse events; and 5) assess the safety of newly licensed vaccines. Results: During 1991--2001, VAERS received 128,717 reports, whereas >1.9 billion net doses of human vaccines were distributed. The overall dose-based reporting rate for the 27 frequently reported vaccine types was 11.4 reports per 100,000 net doses distributed. The proportions of reports in the age groups <1 year, 1--6 years, 7--17 years, 18--64 years, and >65 years were 18.1%, 26.7%, 8.0%, 32.6%, and 4.9%, respectively. In all of the adult age groups, a predominance among the number of women reporting was observed, but the difference in sex was minimal among children. Overall, the most commonly reported adverse event was fever, which appeared in 25.8% of all reports, followed by injection-site hypersensitivity (15.8%), rash (unspecified) (11.0%), injection-site edema (10.8%), and vasodilatation (10.8%). A total of 14.2% of all reports described serious adverse events, which by regulatory definition include death, life-threatening illness, hospitalization or prolongation of hospitalization, or permanent disability. Examples of the uses of VAERS data for vaccine safety surveillance are included in this report. Interpretation: As a national public health surveillance system, VAERS is a key component in ensuring the safety of vaccines. VAERS data are used by CDC, FDA, and other organizations to monitor and study vaccine safety. CDC and FDA use VAERS data to respond to public inquiries regarding vaccine safety, and both organizations have published and presented vaccine safety studies based on VAERS data. VAERS data are also used by the Advisory Committee on Immunization Practices and the Vaccine and Related Biological Products Advisory Committee to evaluate possible adverse events after vaccinations and to develop recommendations for precautions and contraindications to vaccinations. Reviews of VAERS reports and the studies based on VAERS reports during 1991--2001 have demonstrated that vaccines are usually safe and that serious adverse events occur but are rare. Public Health Actions: Through continued reporting of adverse events after vaccination to VAERS by health-care providers, public health professionals, and the public and monitoring of reported events by the VAERS working group, the public health system will continue to be able to detect rare but potentially serious consequences of vaccination. This knowledge facilitates improvement in the safety of vaccines and the vaccination process. IntroductionThe National Childhood Vaccine Injury Act (NCVIA) (1) of 1986 required health professionals and vaccine manufacturers to report to the U.S. Department of Health and Human Services specific adverse events that occur after the administration of routinely recommended vaccines. Postvaccination adverse events and the time frames in which they must occur to qualify as being reportable under NCVIA are listed in the Reportable Events Table (2). The table is updated periodically as the vaccination schedule changes, new vaccines are introduced, and new vaccine-associated adverse events are identified. Vaccine-associated adverse event reports were previously collected separately by CDC and the Food and Drug Administration (FDA). CDC maintained the Monitoring System for Adverse Events Following Immunization (3) for vaccines administered in the public sector; FDA maintained the Spontaneous Reporting System (4) to accept reports from both the public and private sectors, although it was used primarily by vaccine manufacturers. These systems were replaced by the Vaccine Adverse Event Reporting System (VAERS) on November 1, 1990 (5). Under the joint administration of CDC and FDA, VAERS accepts spontaneous reports of suspected vaccine adverse events after administration of any vaccine licensed in the United States (6--9). Unlike many surveillance systems that monitor a single exposure and its associated outcomes, VAERS monitors multiple exposures (i.e., different vaccines often administered simultaneously in different combinations) and an increasing number of potential outcomes. VAERS accepts spontaneous reports from health professionals, vaccine manufacturers, and the public. Reports are submitted by mail or fax. In 2002, electronic reporting to VAERS through the Internet became available by accessing http://secure.vaers.org/VaersDataEntryintro.htm. All reports, whether submitted directly to VAERS by an individual or by state or local public health authorities or manufacturers, are entered into the VAERS database. Federal regulations require that each manufacturer with a product license from FDA report the following adverse events to VAERS: all spontaneous reports of adverse experiences occurring within the United States, whether serious, nonserious, expected or unexpected, and all serious and unexpected adverse experiences occurring outside of the United States or reported in scientific and medical journals as case reports or as the result of formal clinical trials (10). Data collected on the VAERS form (11) include information regarding the patient, the vaccine(s) administered, the reported adverse event, and the person reporting the event. Federal regulations (10) define serious events as those involving death, life-threatening illness, hospitalization or prolongation of hospitalization, or permanent disability. All reports with adverse events classified as serious are followed up with a request for additional information (e.g., medical records and autopsy reports) to provide a complete description of the case. For all original and follow-up reports, the signs, symptoms, and diagnoses mentioned in the description of the adverse event are coded using FDA's Coding Symbols for Thesaurus of Adverse Reaction Terms (COSTART) (12). All information is stored in a computerized database for subsequent reference and analyses. All reporters receive written acknowledgment of receipt of their reports along with a request for missing information where indicated. In addition, letters to obtain information regarding the recovery status of persons with serious adverse events are mailed to the reporters at 60 days and 1 year after vaccination. All personal identifying information is kept confidential as required by law. Medical records submitted to VAERS spontaneously or as part of follow-up activities are also protected by confidentiality requirements. VAERS data stripped of personal identifiers are available at http://www.vaers.org. The primary objectives of VAERS are to 1) detect new, unusual, or rare vaccine adverse events; 2) monitor increases in known adverse events; 3) determine patient risk factors for particular types of adverse events; 4) identify vaccine lots with increased numbers or types of reported adverse events; and 5) assess the safety of newly licensed vaccines. Although VAERS can rarely provide definitive evidence of causal associations between vaccines and particular risks, its unique role as a national spontaneous reporting system enables the early detection of signals (13) that can then be more rigorously investigated. In vaccine safety surveillance, sensitivity takes precedence over specificity. VAERS seeks reports of any clinically important medical event that occurs after vaccination, even if the reporter cannot be certain that the event was caused by the vaccine. The purpose of this report is to provide health-care providers, public health professionals, vaccine manufacturers, and members of the public who are interested in vaccine safety with an overview of the information collected in VAERS regarding adverse events reported during the previous 11 years. Specific examples of how the information was used to assess the safety of the vaccines and how VAERS detected signals that were later followed up are also included. Characterization of reporting profiles for different types of adverse events and vaccines also provides a context within which new and unexpected adverse events reported to VAERS can be interpreted. MethodsThe automated data in the VAERS database were used for analysis. All data were analyzed by using SAS® program version 8 (14). Unless otherwise indicated, only reports received from January 1, 1991, through December 31, 2001, were included. All known duplicate reports (reports concerning the same patients but from different reporting sources) were excluded. All adverse events in the VAERS database were coded using COSTART (12). Reports typically involve multiple COSTART coding terms. Serious adverse events were defined by the federal regulatory definition for seriousness (10), which includes information regarding whether the patient died, experienced life-threatening illness, required hospitalization, and whether the condition resulted in prolongation of hospitalization or in permanent disability. The numbers of adverse event reports in each of the 50 states were calculated by year. The average reporting rates (reports per 1 million population) for each state were calculated by dividing the averages of 11 annual reports of each state by the averages of 1990 and 2000 state population data from the Bureau of the Census. The vaccine-specific reporting rates for each vaccine type (number of reports per 100,000 net doses distributed) were calculated by dividing the number of vaccine-specific reports by the net doses distributed in the United States, according to the data provided by the CDC Biologics Surveillance System (personal communication, Lisa Galloway, National Immunization Program, 2002) (Table 1). These data were provided by the majority of vaccine manufacturers by type of antigen and year of distribution. These net distribution figures are only estimates and serve as approximate denominators for reporting rates of adverse events in the absence of data regarding actual number of doses administered. Net distribution figures represent the total doses distributed by vaccine type during the period, less returned doses. The reporting rates must not be interpreted as incidence rates because whether the vaccine caused the adverse event was uncertain. The adverse event might have occurred by chance after vaccination. In addition, substantial and variable underreporting occurs, and uncertainty exists regarding the actual number of doses administered. The numbers of adverse event reports were calculated in five age groups: <1 year, 1--6 years, 7--17 years, 18--64 years, and >65 years. The unknown age group was defined as not being able to determine age because of missing information. The frequently reported vaccine types or vaccine combinations were defined as vaccine types or vaccine combinations for which >50 adverse event case reports were received. The frequently reported adverse events were defined as the COSTART coding terms of adverse events that were reported >100 times. ResultsSummary of VAERS DataGeneral From January 1, 1991, through December 31, 2001, VAERS received 128,717 case reports describing adverse events after immunization. This report includes data regarding the distribution of these reports by year and the population-based reporting rates in the 50 states (Table 2). The reporting rates varied from 27.7 (Alabama) to 113.2 (Alaska) reports per million population. The four most populous states in the United States (California, Florida, Texas, New York) had low reporting rates of 28.4, 30.3, 32.0, and 35.8, respectively. In contrast, the states with the highest reporting rates were Alaska (113.2), Idaho (81.4), and Wyoming (75.2), which are some of the least populated states. Data regarding the number of adverse event reports for each of the 27 frequently reported vaccine types are included in this report (Table 3). During 1991--2001, >1.9 billion net doses of human vaccines were distributed (Table 1), resulting in an overall dose-based reporting rate for the 27 vaccine types of 11.4 reports per 100,000 net doses distributed. The influenza vaccine (FLU) had the highest distribution (>500 million doses) but the lowest overall reporting rate (3.0 reports per 100,000 net doses distributed). Hepatitis B (HEP) vaccine had the second highest distribution (>200 million net doses) but an overall reporting rate of 11.8 reports per 100,000 net doses distributed. Rhesus rotavirus vaccine-tetravalent (RRV-TV) had the highest overall reporting rate for a specific vaccine (156.3 reports per 100,000 net doses distributed). Two major vaccine substitutions occurred during the 11-year period: diphtheria and tetanus toxoids and acellular pertussis (DTaP) replaced diphtheria and tetanus toxoids and pertussis vaccine (DTP), and inactivated poliovirus vaccine (IPV) replaced oral poliovirus vaccine live trivalent (OPV) for routine vaccinations. The overall reporting rate has decreased substantially after vaccination with DTaP (12.5 reports per 100,000 net doses distributed), compared with that for DTP (26.2). A similar, though limited decrease in average reporting rate was also observed after vaccination with IPV (13.1), compared with that for OPV (15.1) after transition from OPV to IPV in 1996. During the 11-year surveillance period, 44.8% of all reports involved children aged <7 years (<1 year: 18.1% and 1--6 years: 26.7%) (Table 4). The recommended vaccination schedules primarily involve these age groups. A total of 32.6% of all reports were for adults aged 18--64 years, and 4.9% concerned adults aged >65 years. Among children, the difference in sex was minimal in all age groups (<1 year, 1--6 years, and 7--17 years) (Figure 1). In contrast, an excess of reports for women was noted for all adult age groups (18--64 years and >65 years) throughout the surveillance period. Changes in reporting frequencies of different vaccines or vaccine combinations examined by comparing data from two surveillance periods are included in this report (Tables 5 and 6). During the earlier period, 1991--1995, >74% of all VAERS reports mentioned the use of HEP; FLU; measles, mumps, and rubella (MMR); DTP; or tetanus and diphtheria toxoids (Td) vaccines and combined use of DTP with Haemophilus b conjugate virus vaccine (HIBV), OPV, HEP, and MMR (Table 5). Because of the introduction of multiple new vaccines and vaccine combinations and changes in the recommended immunization schedules, the reporting pattern in VAERS changed during the latter period, 1996--2001. Although HEP, FLU, Td, and MMR remained among the most frequently reported vaccines, a substantial number of reports followed the use of varicella (VARCEL), pneumococcal (PPV), anthrax (ANTH), and Lyme disease vaccines (LYME) as well as acellular pertussis vaccines administered either alone or in combination with HEP, HIBV, IPV and/or MMR (Table 6). Overall, the most commonly reported adverse event was fever, which appeared in 25.8% of all reports, followed by injection-site hypersensitivity (15.8%), rash (unspecified) (11.0%), injection-site edema (10.8%), and vasodilatation (COSTART coding term for skin redness) (10.8%) (Table 7). At least one of these primarily nonserious adverse events was mentioned in 74.2% of all VAERS reports. VAERS reports were received primarily from vaccine manufacturers (36.2%), state and local health departments (27.6%), and health-care providers (20.0%), with fewer reports filed directly by patients and parents (4.2%), or others (7.3%) (Table 8). Data documented a continuous increase in the proportion of reporting by health-care providers during the 11-year period. The percentage of reports from health-care providers increased from 11.4% in 1991 to 35.3% in 2001. The improvement in reporting from health-care providers might reflect the efforts of the VAERS working group to enhance communication with physicians through yearly direct mailing, continuing medical education (CME), and other sources. In addition, publications of analyses of VAERS data might have increased health-care providers' recognition of the potential value of reporting. Serious Adverse Events Overall, 14.2% of all reports received in VAERS during 1991--2001 described serious adverse events (10) (Table 9). During 1991--2001, reports of deaths ranged from 1.4%--2.3%, and reports of life-threatening illness ranged from 1.4%--2.8% of all adverse event reports. During the previous 3 years when distribution of vaccines reached the highest level, the annual percentage of reports of death was stable, approximately 1.5% of all adverse event reports. The reports of life-threatening illness were also stable throughout the years except for a peak of 2.8% in 1999, which reflected RRV-TV and intussusception incident that occurred in that year. A clinical research team follows up on all deaths reported to VAERS. The majority of these deaths were ultimately classified as sudden infant death syndrome (SIDS). Analysis of the age distribution and seasonality of infant deaths reported to VAERS indicated that they matched the age distribution and seasonality of SIDS; both peaked at aged 2--4 months and during the winter (15). The decrease in deaths reported to VAERS since 1992--1993 parallels the overall decrease in SIDS in the U.S. population since the implementation of the Back to Sleep campaign (15). Carefully controlled epidemiologic studies consistently have not found any association between SIDS and vaccines (16--19). FDA and the Institute of Medicine (IOM) reviewed 206 deaths reported to VAERS during 1990--1991. Only one death was believed to have resulted from a vaccine. The patient was a woman aged 28 years who died from Guillain-Barré syndrome after tetanus vaccination (20). IOM concluded that the majority of deaths reported to VAERS are temporally but not causally related to vaccination (20). A similar conclusion was reached regarding neonatal deaths temporally reported to VAERS in association with hepatitis B vaccination (21). VAERS in Vaccine Safety SurveillanceIntussusception After Rotavirus Vaccine RRV-TV was licensed in August 1998. The Advisory Committee on Immunization Practices (ACIP) recommendations for its use were published in March 1999 (22). From September 1, 1998, through July 7, 1999, VAERS received 15 reports of intussusception among infants who had received RRV-TV vaccine. CDC reported this finding in July 1999 and recommended that health-care providers postpone use of RRV-TV at least until November 1999, pending results of a national case-control study that was being conducted at that time (23). The manufacturer, in consultation with FDA, voluntarily ceased further distribution of the vaccine in mid-July 1999. On October 22, after a review of scientific data from multiple sources, ACIP concluded that intussusception occurred with substantially increased frequency in the first 1--2 weeks after vaccination with RRV-TV, particularly after the first dose. In 1999, ACIP withdrew its recommendation for vaccination of infants in the United States with RRV-TV (24). From September 1998 through December 1999, VAERS received 121 reports of intussusception among infants who received RRV-TV vaccine (Figure 2). The first intussusception case was reported in December 1998. During the first half of 1999, a total of 14 additional cases of intussusception were reported to VAERS. The majority of cases were reported during July--August 1999, peaking soon after a MMWR publication (July 16, 1999) (23). Other studies have documented similar findings (25--29). All intussusception case-patients reported to VAERS through December 31, 1999, were vaccinated before July 17, 1999 (Figure 3). Before RRV-TV was licensed and marketed in the United States, VAERS had received a total of only three reports of intussusception after other vaccinations (Figure 4). Influenza Vaccine and Guillain-Barré Syndrome Vaccination with swine influenza vaccine is known to increase the risk for Guillain-Barré syndrome (30--34). Reports of Guillain-Barré syndrome after any vaccination are considered serious and followed up by VAERS to obtain additional information. An increase in reports of Guillain-Barré syndrome after the receipt of influenza vaccine was noted in VAERS data by week 29 of the 1993--94 influenza season (35). The number of reports increased from 23 during 1991--92 to 40 during 1992--93 and to 80 during 1993--94 (Figure 5). These findings raised concerns regarding a possible increase in vaccine-associated risk for Guillain-Barré syndrome. A study was initiated to investigate the VAERS signal (35). The study documented that the relative risk of Guillain-Barré syndrome after influenza vaccination, adjusted for age, sex, and vaccine season was 1.7 (95% confidence interval = 1.0--2.8). However, no increase occurred in the risk of vaccine-associated Guillain-Barré syndrome from 1992--93 to 1993--94. For the two seasons combined, the adjusted relative risk of 1.7 indicated that slightly >1 additional case of Guillain-Barré syndrome occurred per 1 million persons vaccinated against influenza. This risk is less than the risk from severe influenza, which can be prevented by the vaccine. In addition, no correlation existed between the number of Guillain-Barré syndrome reports received in VAERS and influenza vaccine doses administered (Figure 5). The annual number of Guillain-Barré syndrome reports has been low and stable during the previous four influenza seasons when the net doses of influenza vaccine distributed increased substantially. This finding reflects data compared with the 1993--94 influenza season in which VAERS received the highest numbers of Guillain-Barré syndrome reports in a single influenza season. This example indicates that VAERS is useful in preliminary evaluation of rare adverse events when the relation to vaccination is uncertain. Safety Assessment After Whole Cell Versus Acellular Pertussis-Containing VaccinesConcerns regarding the safety of DTP vaccines led to a gradual introduction of acellular pertussis-containing vaccines in the United States. In December 1991, FDA licensed the first DTaP vaccine for use in the United States (36). Shortly thereafter, a second DTaP formulation was also licensed (37). Both DTaP vaccines were licensed for use only as the fourth and fifth doses of the DTP series recommended for children aged 15 months--7 years. In July 1996, FDA approved the first DTaP vaccine for infants (38). VAERS reports from 1991 (when whole cell pertussis vaccines were used exclusively) through 2001 (when acellular pertussis vaccines were used predominantly) documented that the overall vaccine-specific reporting rates of both serious and nonserious reports for DTaP had decreased to less than one half of that for DTP among children aged <7 years (Table 10). In comparison with all whole cell pertussis-containing vaccines (DTP and DTPH), the overall nonserious adverse events reporting rate for DTaP vaccines was approximately 40% lower (10.5 versus 16.8 reports per 100,000 net doses distributed). Although reduction in adverse reporting rates is suggestive of a safer vaccine, such comparisons must be interpreted cautiously because reporting rates cannot be viewed as incidence rates. Two studies have documented an improved safety profile of DTaP vaccines based on review of VAERS data from 1991--1993 among children and 1995--1998 among infants (39,40). The decreasing trends for selected systemic adverse events (e.g., fever) and neurologic reactions (e.g., seizures) continued to be observed during 1999--2001 (Figures 6 and 7). However, an increase in the number of reports concerning injection-site reactions was detected by the end of this surveillance period (Figure 8). The increase is more prominent among the recipients of booster doses of DTaP (fourth and fifth dose). This finding is consistent with the results of a recent study that documented an increase in the risk of extensive local reactions in recipients of fourth and fifth doses of the DTaP vaccines (41). Safety Assessment After IPV Versus OPVSince it was licensed in 1963, OPV has been the vaccine used for the prevention of poliovirus infection in the United States. The use of OPV led to the elimination of wild-type poliovirus in the United States in <20 years. However, the risk of vaccine-associated paralytic poliomyelitis (VAPP) was estimated to be approximately 1 case per 2.4 million doses distributed, with the majority of VAPP cases occurring after the administration of the first dose (1 case per 750,000 first doses) (42,43). The reporting sensitivity of VAPP in VAERS was an estimated 68%--72% (44). In September 1996, to reduce the occurrence of VAPP, ACIP recommended an increase in the use of IPV through a sequential schedule of IPV followed by OPV (42). VAERS has not received any report of VAPP after OPV/IPV vaccination since 1997, suggesting a positive effect of the sequential schedule of IPV followed by OPV (Figure 9). This result is consistent with previously reported data (45). In July 1999, ACIP recommended that IPV be used exclusively in the United States to maintain disease elimination and to prevent any further cases of VAPP (46). Safety Assessment After Varicella VaccineIn March 1995, varicella vaccine was licensed in the United States. In July 1996, varicella vaccine was recommended by ACIP for all children without contraindications at aged 12--18 months, for all susceptible children by their thirteenth birthday, and for susceptible adolescents and adults who are at high risk for exposure to varicella (47). In February 1999, ACIP expanded its recommendations for varicella vaccine to promote an expanded use of the vaccine for susceptible children and adults (48). VAERS received 15,180 adverse event reports after varicella vaccination from March 1995 through December 2001, the majority (14,421, or 95%) of which described nonserious events. The highest numbers of reports were received soon after licensure (Figure 10). As the net distribution of varicella vaccine increased, the number of adverse event reports decreased continuously over the years. Of the 15,180 adverse event reports received, the number of serious adverse events reported for varicella vaccine was 759 (5%). The proportion of reports of serious adverse events was stable over the years (range: 3.7%--6.3%). A detailed review of VAERS reports received during the first 3 years after the licensure of varicella vaccine documented that the majority of reported adverse events for varicella vaccine were minor, and serious events were rare (49). A vaccine etiology for the majority of reported serious events could not be confirmed; further research is needed to clarify whether varicella vaccine played a role. Safety Assessment After Lyme Disease VaccineIn December 1998, FDA licensed the first vaccine to prevent Lyme disease. ACIP stated that the vaccine should be considered for persons who reside in areas where Lyme disease is endemic and who have frequent or prolonged exposure to tick-infested habitats (50). Review of early reports to VAERS revealed adverse events that corresponded to Lyme vaccine safety data from the prelicensure trials, including injection-site reactions, transient arthralgia and myalgia within 30 days of vaccination, fever, and flu-like symptoms. Hypersensitivity reactions, not observed in the clinical trial, were also reported to VAERS. Some of the reported hypersensitivity reactions can be linked to the vaccine on the basis of the specificity of the symptoms, close temporal proximity to vaccination, and the known association of the reactions with other vaccines. For other reported adverse events, causal relations with Lyme disease vaccine have not been established. No clear patterns in age, sex, time to onset, or vaccine dose have been identified. The onset of symptoms consistent with Lyme disease (e.g., facial paralysis and arthritis) after Lyme disease vaccination has also been reported to VAERS. Determining whether the facial paralysis was part of the expected background incidence or attributable to the vaccine or to Lyme disease was not possible. A higher proportion of arthritis-related events was reported after the second or third dose compared with all events combined. This higher proportion might be attributable to the increased amount of time available for a vaccine recipient to report an adverse event: 11 months between the second and third doses (51). Because of persistent public concerns, a follow-up study was conducted to further evaluate reports of arthritis after vaccination for Lyme disease. In 7 of 14 confirmed arthritis cases, a history of concomitant exposure or another medical condition existed, including Lyme disease, that provided a possible explanation for arthritis (52). In early 2001, the manufacturer withdrew the vaccine from the market, citing poor sales. DiscussionThis report provides an overview of reports to VAERS during 1991--2001. The VAERS data should be interpreted with caution, because they describe events that occurred after vaccination but they do not necessarily imply that the events were caused by vaccination. Although the 128,717 adverse event reports received in VAERS during the previous 11 years are a substantial number, it is low in comparison with the >1.9 billion doses of vaccines administered in the United States during the same period (Table 1). VAERS seeks to capture as many clinically important medical events after vaccination as possible, even if the person who reported the event was not certain that the incident was vaccine-related. Temporal association alone does not mean that the vaccine caused the illness or symptoms. The illness or symptoms could have been a coincidence or might have been related to an underlying disease or condition or might have been related to medicines or other products taken concurrently. During 1999--2001, more reports were submitted to VAERS annually than in the early 1990s. Multiple factors that likely contributed to this increase include the introduction of new vaccines in the mid- to late 1990s (rotavirus vaccine, Lyme disease vaccine, varicella vaccine, and pneumococcal conjugate vaccine), the increased use of anthrax vaccine by military personnel, and the increase in the number of doses of vaccines administered to both adult and children (Table 1). In addition, reporters have become increasingly aware of VAERS. Because of the diverse population VAERS covers and the number of reports it receives, VAERS is useful for detecting new, unusual, or rare events and assessing newly licensed vaccines. Review of reports during the initial months of licensed use of a new vaccine cannot only rapidly identify problems not detected during prelicensure evaluation (e.g., intussuception and RRV- TV) but also reassure the general public concerning the safety of a new vaccine, as in the safety assessments of varicella vaccine and hepatitis A (HEPA) vaccine (53). VAERS has also been useful in screening for unusual increases in previously reported adverse events (e.g., influenza vaccine and Guillain-Barré syndrome investigation during the 1992--93 and 1993--94 influenza seasons). Investigating changes in reporting rates in VAERS might lead to positive change in vaccine practices. After the licensure of DTaP for the fourth and fifth doses in the vaccination schedule of older children, VAERS data were used to compare reporting rates for specific adverse events after DTaP versus DTP within the first 72 hours after vaccination (39). This study confirmed a better safety record for DTaP among older children and was one factor in ACIP's subsequent recommendation for the use of DTaP among infants. As was also critical in the safety assessment of IPV versus OPV, VAERS provided evidence of improved safety in evaluating changes in immunization practices recommended by ACIP. VAERS has also facilitated the lot-specific safety evaluations, which have periodically been of public concern. Lot sizes vary substantially. Every lot of vaccine must meet strict criteria for purity, potency, and sterility before it can be released to the public by the manufacturer. FDA medical officers review all reports of death and other serious events, and they also look each week for clusters within the same vaccine lot. In addition, FDA medical officers evaluate reporting rates of adverse events by lot, as needed, looking for unexpected patterns. During the 11 years, no lot needed to be recalled on this basis. VAERS is subject to the limitations inherent in any passive surveillance system (54). Among those, underreporting (only a fraction of the total number of potentially reportable events occurring after vaccination are reported) and differential reporting (more serious events and events with shorter onset time after vaccinations are more likely to be reported than minor events) are most noticeable (44). Overreporting also occurs because certain reported adverse events might not be caused by vaccines, and some reported conditions do not meet standard diagnostic criteria. Many reported events, including serious ones, might occur coincidentally after vaccination and are not causally related to vaccination. Other potential reporting biases include increased reporting in the first few years after licensure, increased reporting of events occurring soon after vaccination, and increased reporting after publicity about a particular known or alleged type of adverse event. Individual reports might contain inaccurate or incomplete information. Because of all of these reasons as well as the absence of control groups, differentiating causal from coincidental conditions by using VAERS data alone usually is not possible. Other methodologic limitations of VAERS include the fact that it does not provide information regarding background incidence of adverse events in the general population nor does it provide information concerning the total number of doses of vaccine or vaccine combinations actually administered to patients. Despite its limitations, VAERS contributes to public health in critical ways. CDC and FDA have published and presented numerous vaccine safety studies based on the analyses of VAERS data (55). The high number of reports and the national coverage increase the possibility of detecting or better understanding adverse events that might occur too rarely to be considered as a signal in prelicensure clinical trials or even in a postmarketing active surveillance program. The identification of signals by monitoring VAERS data might initiate further investigation of potential problems in vaccine safety or efficacy and subsequent dissemination of safety-related information to the scientific community and the public. VAERS is also used to evaluate the safety of vaccines used in unique populations (e.g., travelers and the military). Studies have been published regarding Japanese encephalitis (56), Lyme (51), meningococcal (57), and yellow fever vaccines (58,59), among others. To provide a more rigorous setting in which investigators can follow up on signals from VAERS or concerns arising from other sources, the Vaccine Safety Datalink (VSD) Project, a large-linked database, was established in 1991 (60). VSD includes information concerning >7 million persons in eight health maintenance organizations (HMOs) throughout the United States. The strengths of VSD include the documentation of immunizations, the absence of underreporting bias of medical outcomes, and the inclusion in the database of a high number of vaccinated persons who did not have adverse events. However, the VSD data are not available for analysis in as timely a manner as the VAERS data and are not fully representative of the U.S. population regarding race, socioeconomic status, health-care setting, or vaccine lot uses. Nonetheless, VSD permits the conducting of planned epidemiologic vaccine safety studies as well as, in certain situations, urgent investigations of new hypotheses (28). In addition to VSD, CDC has established a new collaborative project, the national network of Clinical Immunization Safety Assessment (CISA) Centers. The centers will develop and disseminate standardized clinical evaluation protocols to clinicians. In addition, the CISA centers will provide referral and consultation services to health-care providers regarding the evaluation of patients who might have had an adverse reaction to vaccination, which will include how to manage the adverse reaction and provide counsel on advisability of continued vaccination. The CISA centers will undertake outreach and educational interventions in the area of vaccination safety. The objectives of CISA are to enhance understanding of known serious or unusual vaccine reactions, including the pathophysiology and risk factors for such reactions, as well as to evaluate newly hypothesized syndromes or events identified from the assessment of VAERS data to clarify any potential relation between the reported adverse events and immunization. Certain adverse events are rarely seen in clinical trials, and clinicians see them too rarely to manage them in a standardized manner. CISA will fill this gap by assisting clinicians in the management of adverse events after immunization. Acknowledgments The authors acknowledge the contributions of the other members of the VAERS working group, Scott Campbell, M.P.H., Kathleen Fullerton, M.P.H., Sharon Holmes, Young Hur, M.D., Elaine Miller, M.P.H., Susanne Pickering, M.S., and Ali Rashidee, M.D., National Immunization Program; Dale Burwen, M.D., David Davis; Phil Perucci, Sean Shadomy, D.V.M., Frederick Varricchio, M.D., P.h.D., and Jane Woo, M.D., Food and Drug Administration, Rockville, MD; and Vito Caserta, M.D. and Geoffrey Evans, M.D., Health Resources and Services Administration, Rockville, Maryland. We also acknowledge Stephen Gordon, Pharm.D. and other staff of Analytical Sciences, Inc., Durham, North Carolina; Xiaojun Wang, M.D., Emory University Rollins School of Public Health, Atlanta, Georgia; and John Grabenstein, MD, Department of Defense, Washington, D.C. In addition, the authors acknowledge Walter Orenstein, M.D., Susan Chu, Ph.D., Mary McCauley, MTSC, Benjamin Schwartz, M.D., and Phil Smith, Ph.D., National Immunization Program for their review of the manuscript; and the health-care providers, public health professionals, and members of the public who have reported events of potential concern to VAERS. References
Vaccine Codes* Used in the Vaccine Adverse Event Reporting System (VAERS) Return to top. Table 1 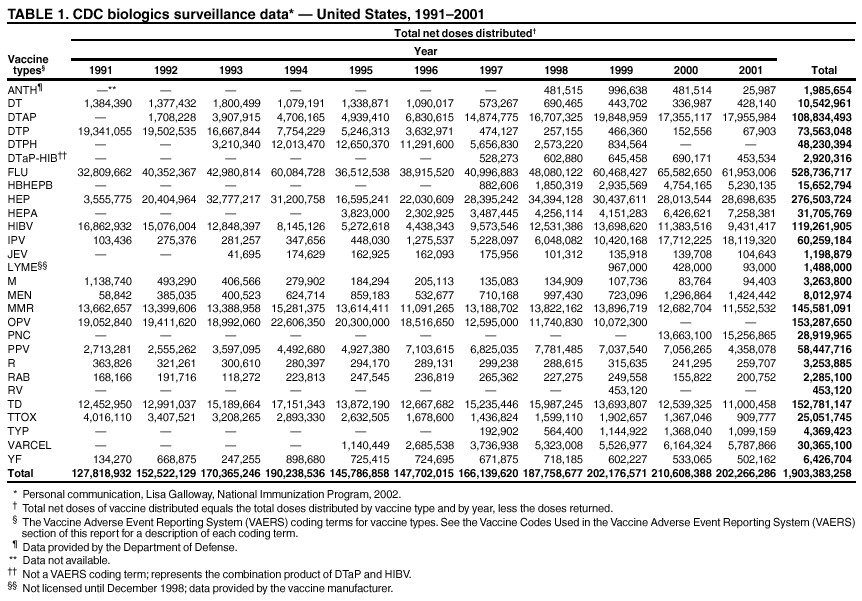 Return to top. Figure 1 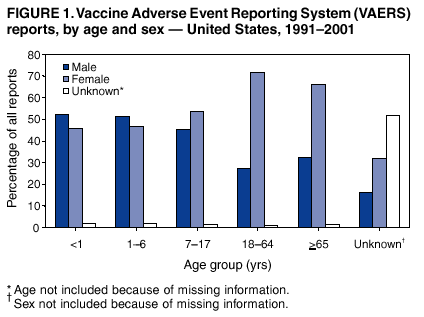 Return to top. Table 2 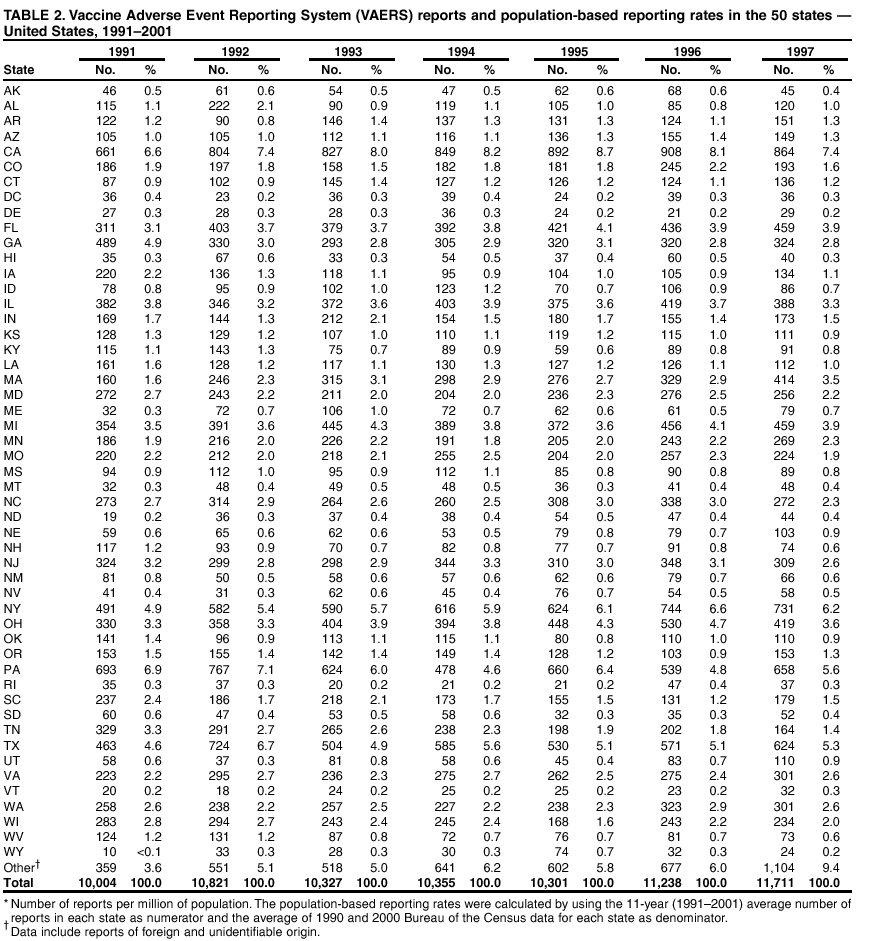 Return to top. 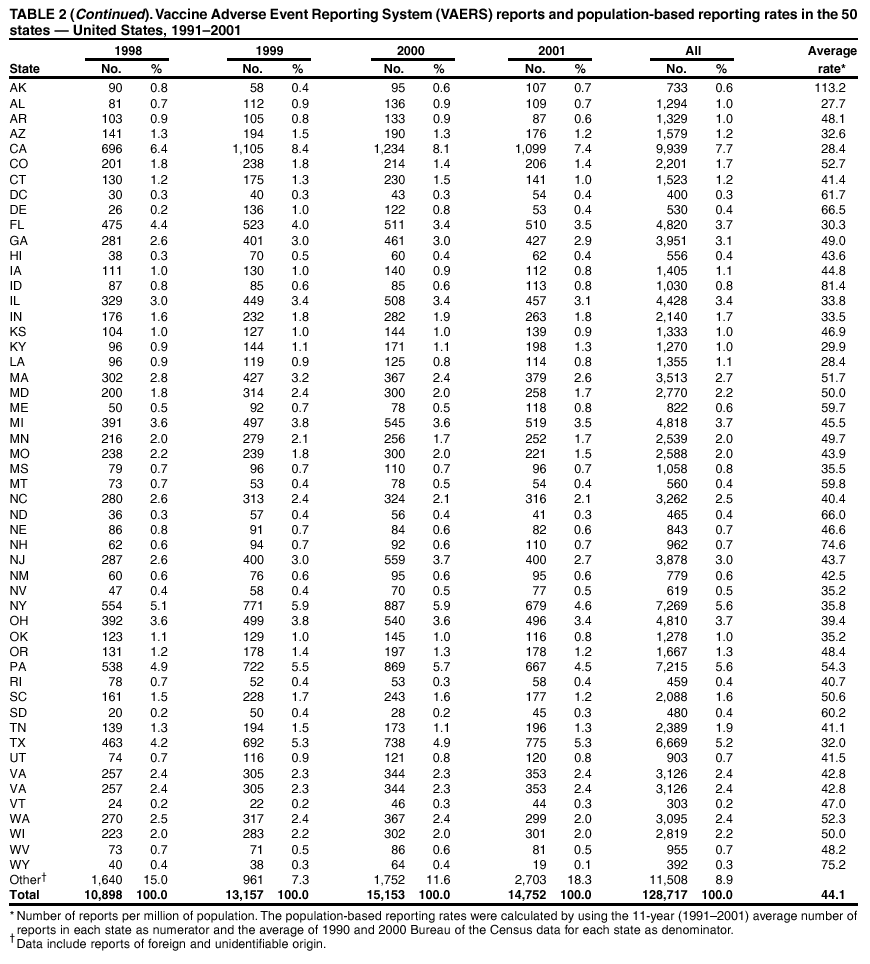 Return to top. Figure 2 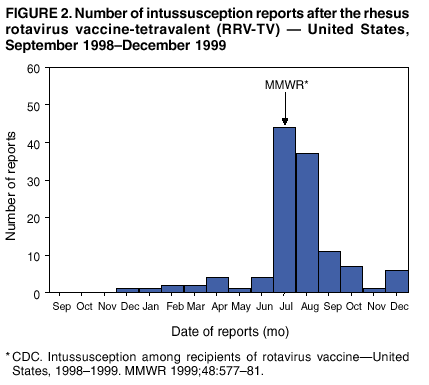 Return to top. Table 3 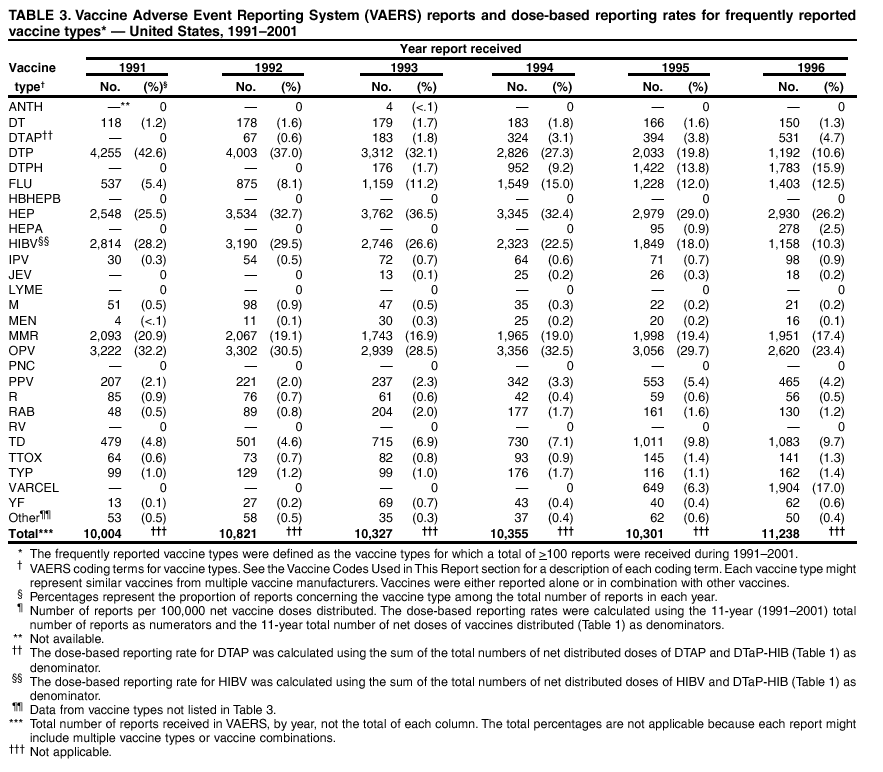 Return to top. 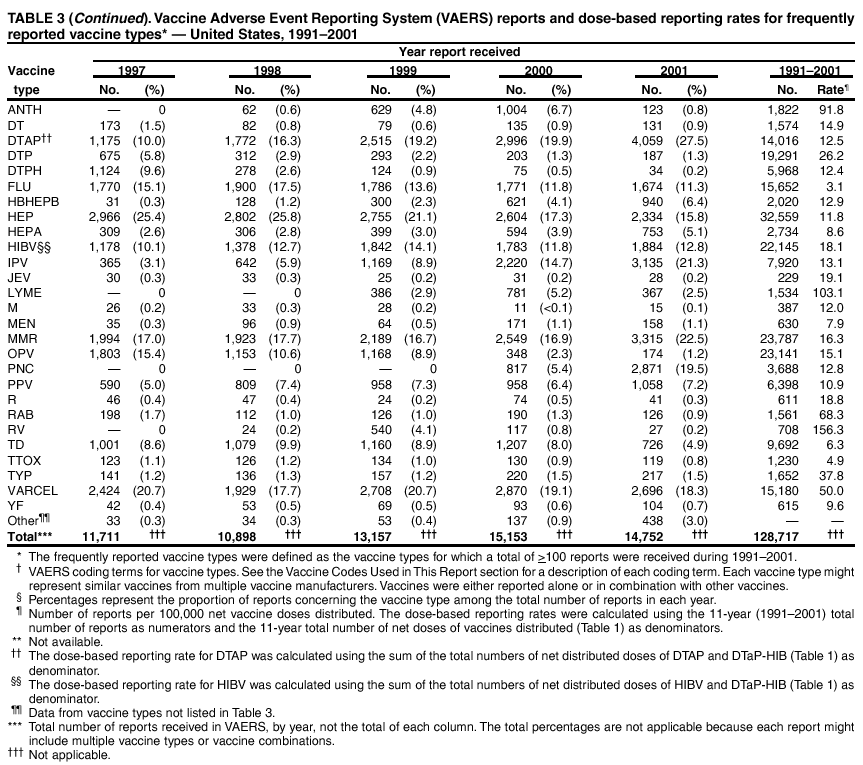 Return to top. Figure 3 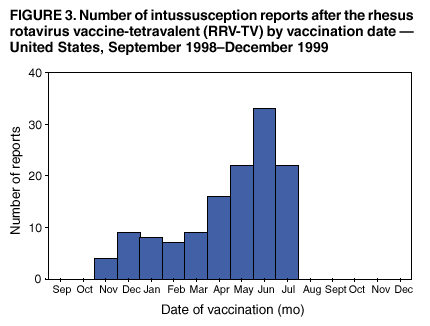 Return to top. Table 4 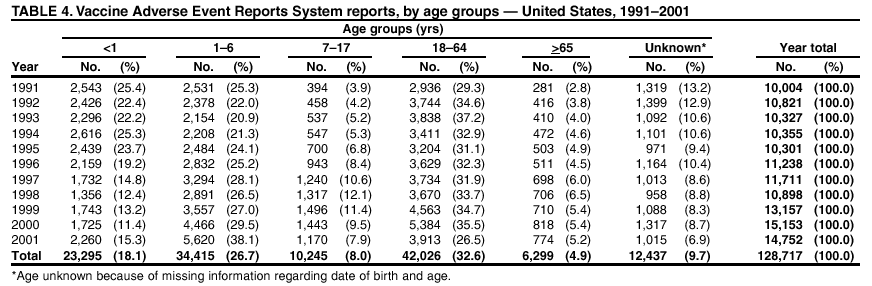 Return to top. Figure 4 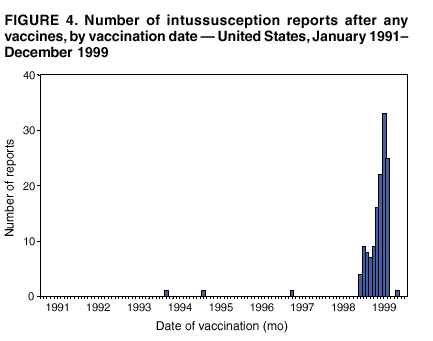 Return to top. Table 5 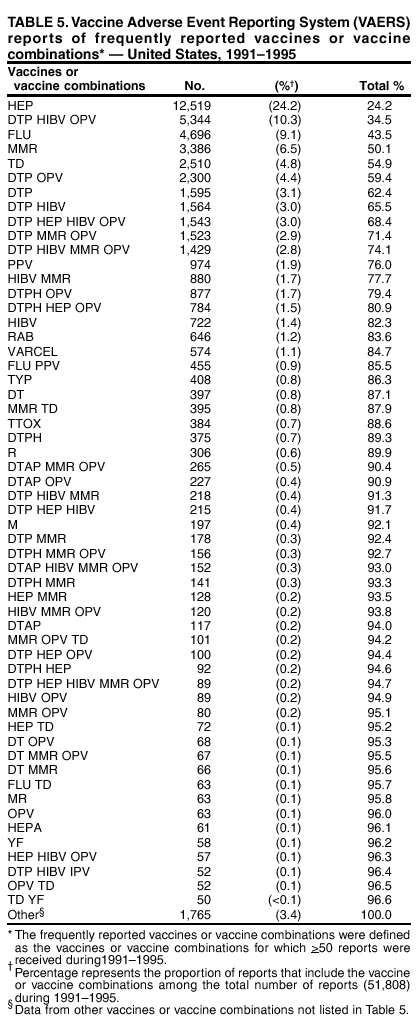 Return to top. Figure 5 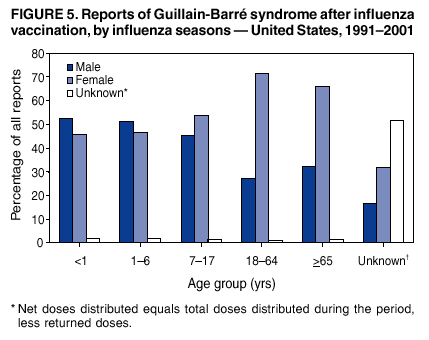 Return to top. Table 6 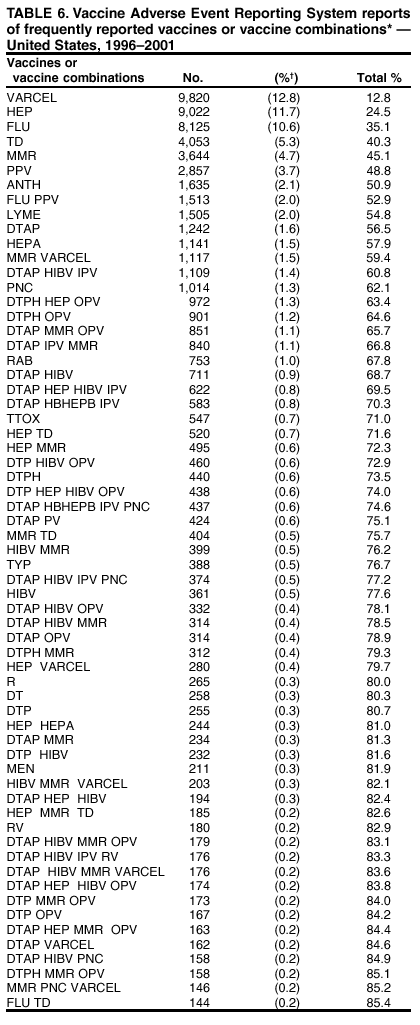 Return to top. 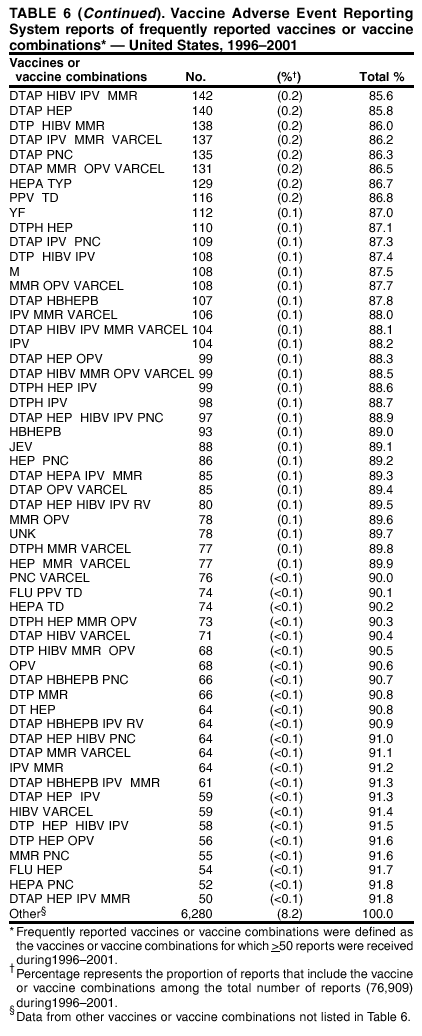 Return to top. Table 7  Return to top. 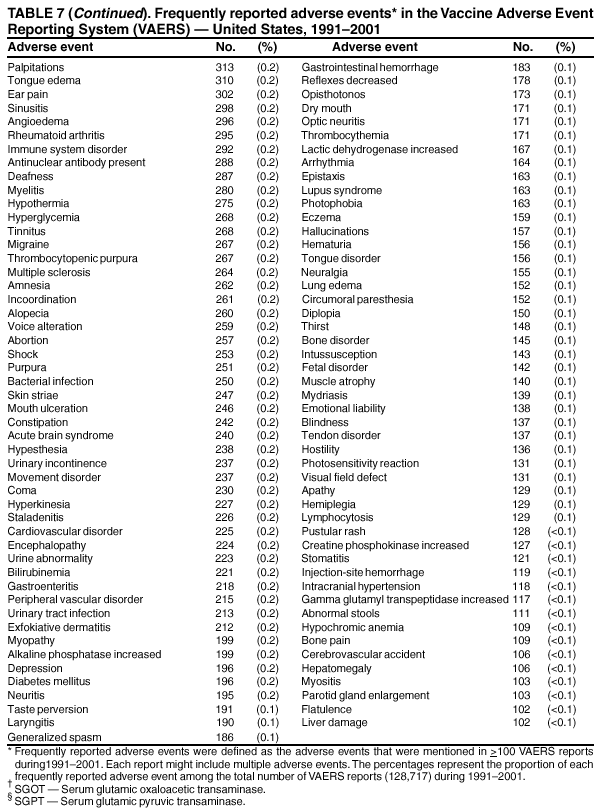 Return to top. Table 8 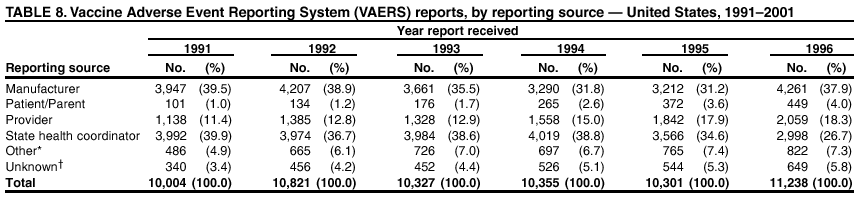 Return to top. 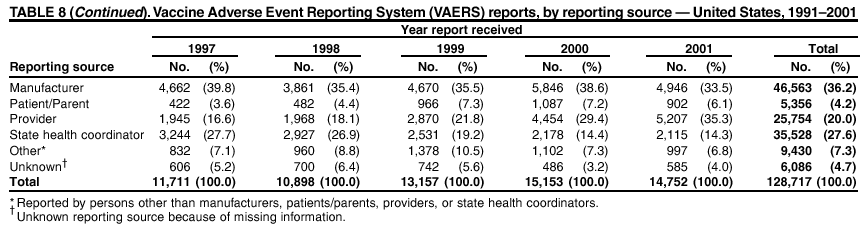 Return to top. Figure 8 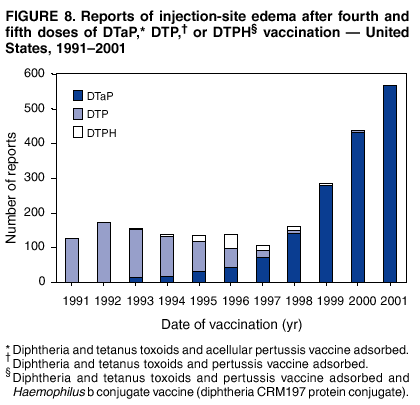 Return to top. Table 9 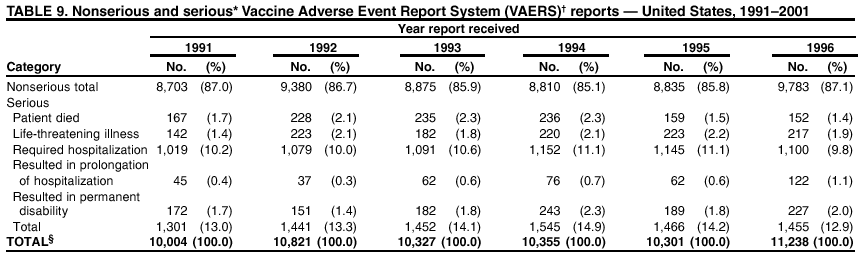 Return to top. 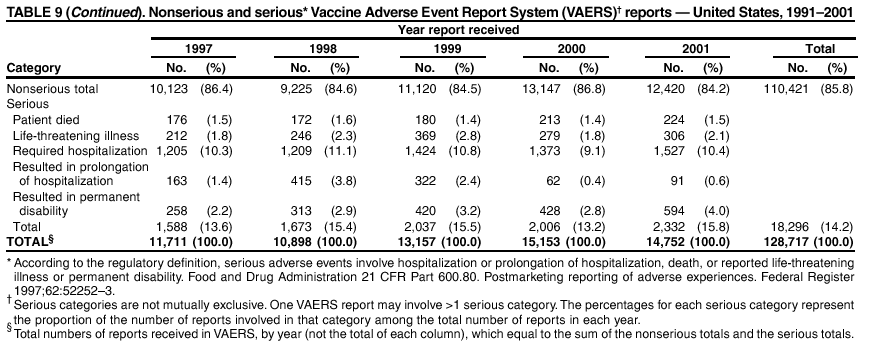 Return to top. Figure 9 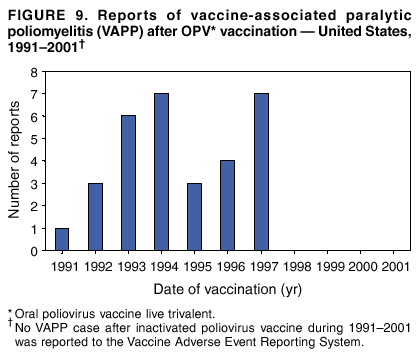 Return to top. Table 10 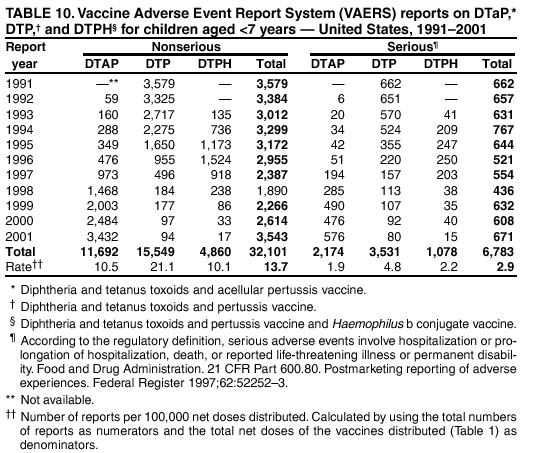 Return to top. Figure 10 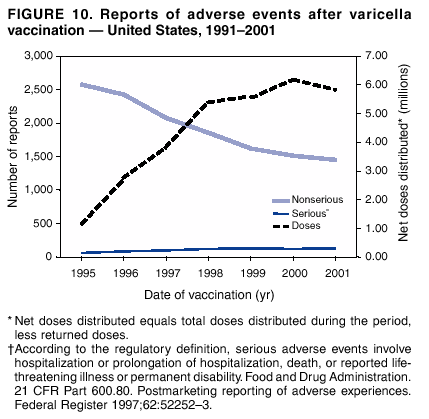 Return to top.
Disclaimer All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to [email protected].Page converted: 1/8/2003 |
|||||||||
This page last reviewed 1/8/2003
|